You're in the right place. ChemScan and Partech are now part of In-Situ, and you'll find information on all products here.
In-Situ Process
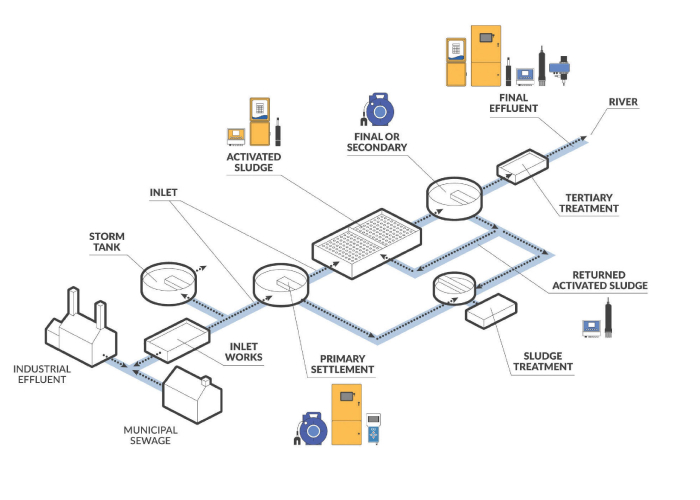
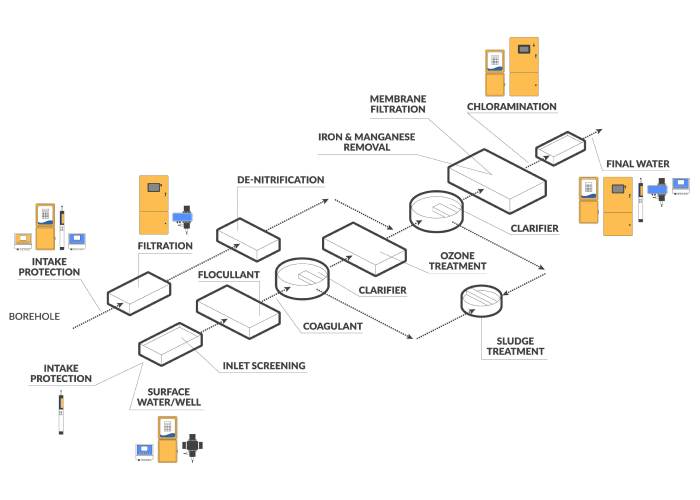
Simplify data collection and access at every stage of your process with industrial-grade online analyzers, in-tank sensors, portable monitors and flexible communication options.
Online Analyzers
Reliable and easy-to-use single-line and multiple-line water analyzers are renowned for low-maintenance operation, accurate analysis and real-time data.
A full line of low-maintenance digital sensors is designed to perform in harsh environments for measurement of dissolved oxygen, pH, redox/ORP, conductivity, color, temperature, salinity, suspended solids and turbidity.
The MPX4 Multiparameter Sonde, with interlocking, interchangeable sensors, anti-fouling wiper and Bluetooth® communication, is ideal for deployed or handheld water quality monitoring.
VuLink cellular telemetry, with one-press setup and enhanced reliability, simplifies remote water quality monitoring and makes it easy to access and manage data from anywhere.